

Five years ago, a team at Google’s DeepMind won CASP14, the long-running protein structure prediction competition, with AlphaFold, a machine-learning system capable of predicting the three-dimensional structures of many proteins at near-experimental accuracy. The advance fundamentally reshaped structural biology and was recognized with a Nobel Prize in 2024.
The success of AlphaFold has been transformative, but it has also made clear what remains missing: the chemistry-level accuracy needed for drug design and a grasp of how proteins change shape as they interact with other proteins, hormones, metabolites, and their physical environment. The scale of experimental data collection has only increased since AlphaFold, while structure determination has emerged as the primary bottleneck. Each of these atomic structures still requires days or weeks of human effort to build. More precisely, it takes extensive human effort to build atomic models that accurately represent the underlying imaging or scattering data.
“We got very excited about the possibility of using all the knowledge baked into models like AlphaFold to help us interpret our experimental data,” says Doeke Hekstra, MCB associate professor. “The catch was, no machine learning model was actually capable of directly interacting with the underlying data—only with atomic models of the data.” Applied Physics graduate student Minhuan Li in Hekstra’s group set out to develop the missing tool, SFCalculator, a GPU-enabled, end-to-end differentiable mapping between atomic models and the underlying cryo-EM, cryo-ET, or X-ray data, which they describe in a recent preprint.
With this tool in hand, the team began speaking with Alisia Fadini, then a postdoc working with Randy Read and Airlie McCoy at the University of Cambridge, as well as with Mohammed AlQuraishi at Columbia University, and realized they were on the same path. AlQuraishi’s group had created the missing piece: OpenFold, an open-source version of AlphaFold. “Once Alisia and Minhuan put these pieces together, their progress was breathtaking.”
The result of over two years of intensive collaboration is ROCKET—an extension of OpenFold that can modify its internal representation of protein evolution in response to experimental data. In work now published in Nature Methods (PDF) , the team shows how ROCKET can handle a wide array of experimental data. “Although we started with X-ray crystallography, it has been stunning to see just how well the approach extended to cryo-electron microscopy and tomography,” says Hekstra. “We see that the prior information encoded by AlphaFold is especially helpful in interpreting low-resolution cryo-EM and ET data. That’s where ROCKET really shines.”
It was already known that the evolutionary record of protein sequences contains information about the different shapes proteins can adopt, but it was not obvious how one could get AlphaFold to use that information effectively. “We found that ROCKET can jump from one plausible conformation to another by stochastic gradient descent,” adds Hekstra. “It is quite striking to watch ROCKET as it searches: the jumps it makes are unlike anything most traditional methods would do.”
Movie 1:

ROCKET’s structure refinement (pink) of a c-Abl kinase structure (pink), flipping the kinase activation loop quickly and adjusting the remainder of the protein. Grey: deposited PDB structure shown as a reference.
Movie 2:
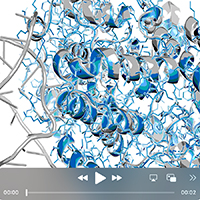
ROCKET’s refinement of a time-resolved snapshot of DNA photolyase, a light-dependent DNA repair protein. The refined model is colored according to the value of the experimental loss (lower = better fit to the data). Grey: deposited PDB structure shown as a reference.
Using new datasets from collaborators Amir Khan (Trinity College, Dublin) and Luca Jovine (Karolinska Institute, Stockholm), the team showed that new insights, supported by complementary data, could now be obtained from very challenging cryo-EM and ET datasets.
Using ROCKET does not require deep expertise in structural biology (see this webinar): The method will be available in several ways: integrated into PHENIX, the widely used structure-determination software platform (forthcoming), distributed through Harvard’s SBGrid, a platform that delivers ready-to-use structural biology tools to academic and industry researchers around the world, and through RS-Station, an open-source software development platform and community that originated in the Hekstra lab.
![]() (PDF)
(PDF)


