

As our brain develops, neurons need to connect to each other through synapses. We study a family of proteins called clustered protocadherins that help ensure that only proper neuronal connections are formed. The term “clustered” refers to the genomic organization of this large protein family: our 53 clustered protocadherin genes are clustered together on one region of the same chromosome. A clustered protocadherin is a membrane protein with an extracellular domain that specifically pairs with another copy of itself on a neighboring neuron, thus forming “homodimers.” The formation of these homodimers is key to proper synapse formation, thus evolution has generated this large collection of isoforms to have exquisite specificity, that is, a near-absolute preference to form homodimers over heterodimers. How did evolution do it?
In a previous study, we elucidated the structure of a clustered protocadherin homodimer (see figure), confirming our earlier bioinformatic prediction of an extended antiparallel dimer interface in collaboration with Debora Marks’s lab (Harvard Medical School). We analyzed protein sequences for diversification and coevolution to investigate how isoform self-specificity—formation of homodimers—is determined. We inferred that different protein interface regions on the large dimer interface are responsible for either the strength or the specificity of the homodimer interaction.
In our new paper (PDF), we used the clustered protocadherins as a case study to develop new tools and concepts of how nature achieves molecular specificity between a large number of paralogs in a single genome. Continuing our collaboration with the Marks lab, we built a statistical model of molecular specificity in the clustered protocadherin family. A key feature of this model is that it can be used to assess likelihood of interaction for sequence pairings not yet seen in nature, which allows us to compute likelihood of interaction for all possible pairs of isoforms, and to assess the effect of residue mutations at the interface. We used this model to assess the contribution of individual regions to the interaction specificity, generally confirming but also identifying exceptions to our earlier findings about division of labor between different parts of the interface.
The individual structures of clustered protocadherin homodimers visualized by x-ray crystallography to date suggest that each has its idiosyncrasies, and also that the dimers may be more dynamic than individual static pictures indicate. To test this idea more directly, we teamed up with Marcos Sotomayor’s lab at The Ohio State University to perform molecular dynamics simulations, modeling the protein dynamics in silico. The simulations showed quite large fluctuations in binding interactions of individual domains over time, leading us to propose what may be a general mechanism of specificity in large interfaces: that weak binding of individual domains leads to a strong polyvalent interaction.
We then combined our molecular dynamics simulations with sequence coevolution analyses to show for the first time that highly coevolving residue pairs interact more frequently across a protein-protein interface during dynamic interactions. This finding expands our understanding of the driving forces behind evolutionary couplings—that sites that are more frequently in contact undergo stronger evolutionary pressure. The combination of techniques also adds robustness to the statistical model: pairing molecular dynamics with coevolution gives more insight into a protein family, particularly when there is only one or a few structures available.
Previous studies using sequence coevolution to understand molecular specificity have focused exclusively on bacterial proteins like two-component systems and toxin-antitoxin systems. Our work represents the first application of coevolutionary methods to study the specificity of eukaryotic proteins. This suggests that other high-interest protein families in eukaryotes, such as G-protein coupled receptors, Notch-delta, and insect sensory receptors, each of which contain many family members per genome, would be amenable to similar analyses.
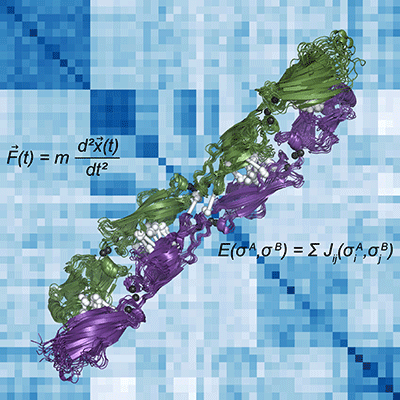
Pictured is a computed-generated representation of the clustered protocadherin homodimer in green and purple. The binding specificity of these proteins is integral to the development of the nervous system in vertebrates. To understand the evolution of this specificity, we used molecular dynamics simulations (the force equation for these simulations in the upper left and the resulting protein dynamics illustrated in the superimposed structures) and sequence coevolution (coevolving residue pairs across the interface shown as white dumbbells) to build a statistical model of interaction specificity (the energy of interaction equation is in the lower right; the resulting heat map of the pairwise statistical energy of interaction values for all clustered protocadherins in the background is colored white to blue for weak to strong predicted interaction).
by Rachelle Gaudet, John M. Nicoludis, and Anna Green



